We are conducting highly original research focused on adaptive immune responses to non-protein antigens, such as glycans and nucleic acids. In parallel, we are developing novel therapeutics for autoimmune diseases as well as therapeutic vaccines aimed at regulating cancer immunity and allergic responses.
Contents
B cell responses to non-protein antigens play important roles in infection immunity and autoimmune diseases
B cells can recognize non-protein antigens such as nucleic acids, polysaccharides, and glycolipids, and produce specific antibodies against them. On the other hand, conventional T cells are unable to recognize non-protein antigens, except for some glycolipids. For this reason, adaptive immune responses to non-protein antigens are mainly mediated by B cells. Antibodies against non-protein antigens play a central role in defense against infections by encapsulated bacteria such as Streptococcus pneumoniae. In autoimmune diseases like Guillain-Barré syndrome—and to some extent in systemic lupus erythematosus (SLE)—the production of autoantibodies targeting non-protein autoantigens such as glycolipids or nucleic acids is implicated in disease onset.
Mechanism of B cell responses to non-protein antigens
Textbooks state that for B cells to produce antibodies, they generally require help from activated T cells that recognize the same antigen. However, T cells are typically unable to recognize non-protein antigens—except for certain glycolipids—and therefore, they generally cannot assist in the activation of B cells that recognize non-protein antigens. Instead, non-protein antigens can provide endogenous signals that contribute to B cell activation. These signals appear to support the activation of B cells specific to the non-protein antigen, enabling the production of antibodies even in the absence of T cell help.
For example, nucleic acids are detected by nucleic acid sensors such as TLR7, which initiate activation signaling pathways. However, the intrinsic activation signals associated with glycan-related antigens, such as polysaccharides and glycolipids, remain poorly understood. Our research group is currently investigating the mechanisms by which polysaccharides activate B cells independently of T cell help.
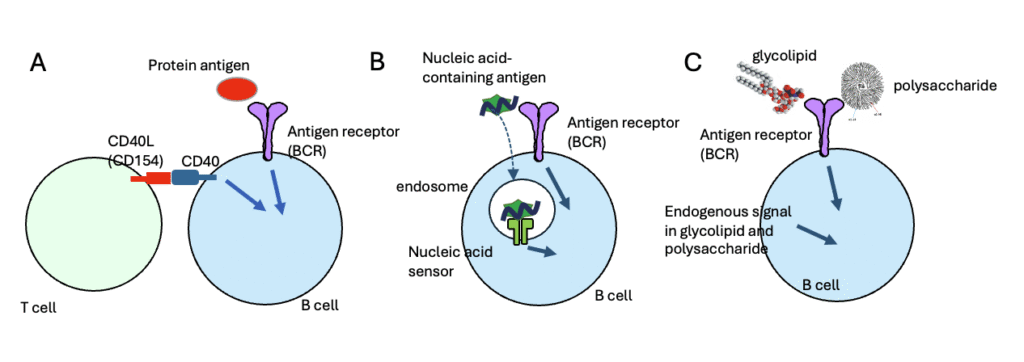
Figure 1. Mechanisms of B Cell Activation by Protein and Non-Protein Antigens
(A) B cell response to protein antigens depends on T cell help. When B cells recognize a protein antigen via the B cell receptor (BCR), BCR signaling is initiated. Subsequently, the B cells interact with T cells that recognize the same antigen. Through this interaction, CD40 ligand (CD40L, also known as CD154) expressed on T cells binds to CD40 on B cells, delivering co-stimulatory signals. BCR signaling alone is insufficient for B cell activation; activation requires both BCR signaling and CD40-mediated co-stimulatory signaling provided by T cell help. (B) T cell-independent activation of B cells by nucleic acid-containing antigens. When B cells recognize nucleic acids or nucleic acid-containing complex antigens, BCR signaling is triggered. These antigens are internalized via BCR-mediated endocytosis, allowing the nucleic acids to engage endosomal sensors such as TLR7 and TLR9. These sensors provide co-stimulatory signals that, together with BCR signaling, lead to B cell activation independently of T cell help. (C) T cell-independent B cell activation by glycolipids and polysaccharides. Glycolipids and polysaccharides possess intrinsic co-stimulatory properties that can directly activate B cells recognizing these antigens. However, the precise molecular nature of these endogenous co-stimulatory signals remains unclear.
Elucidation of the mechanism that prevents autoimmunity to non-protein autoantigens
Our research group has been studying inhibitory receptors expressed in B cells. CD72 is an inhibitory receptor expressed exclusively on B cells and is genetically associated with human systemic lupus erythematosus (SLE) as well as SLE-like autoimmune diseases in mice. Furthermore, both an overseas research group and our own have established CD72-deficient mice and demonstrated that these mice spontaneously develop SLE. These findings indicate that CD72 plays a critical role in suppressing the development of SLE.
In SLE, autoantibodies are produced against various molecular complexes containing nucleic acids, which contribute to disease pathogenesis. Among these, autoantibodies against the Sm/RNP complex and ribosomes are characteristic of SLE, as they are not found in other rheumatic diseases. We have shown that CD72 recognizes Sm/RNP and ribosomes as ligands, thereby inhibiting B cell activation and promoting self-tolerance in B cells that recognize these self-antigens.
Patients with Guillain-Barré syndrome produce autoantibodies against a subset of glycolipids called gangliosides, which are expressed in neurons. These autoantibodies cause peripheral neuropathy. We have demonstrated that the inhibitory receptor Siglec-10, which is expressed in B cells, recognizes various gangliosides. Additionally, we discovered a rare loss-of-function mutation in SIGLEC10 and demonstrated that this mutation is strongly associated with Guillain-Barré syndrome. This finding suggests that Siglec-10 recognizes gangliosides and suppresses the production of ganglioside-specific autoantibodies by B cells, thereby preventing the development of Guillain-Barré syndrome.
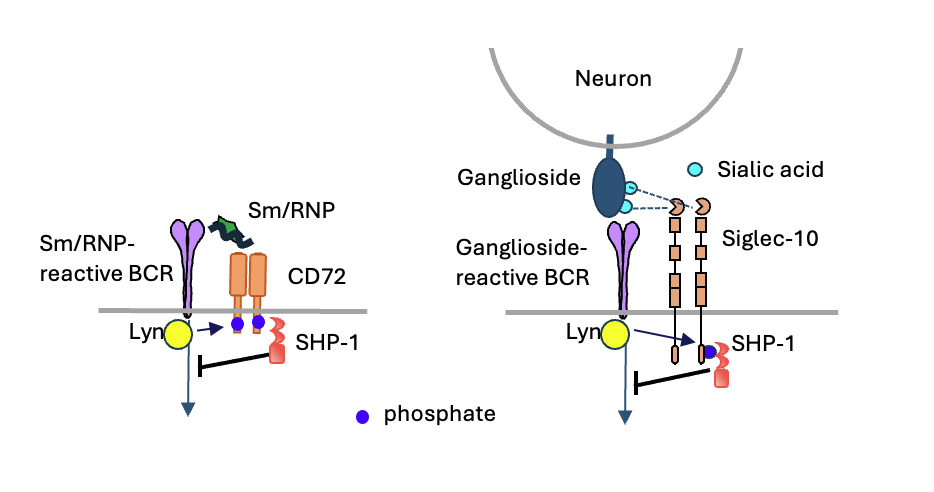
Figure 2. Inhibitory Receptors Induce B Cell Tolerance to Nucleic Acid-Containing and Glycolipid Self-Antigens
B cells express inhibitory receptors such as CD72 and Siglec-10, which promote self-tolerance by suppressing activation of B cells that recognize specific non-protein self-antigens. CD72 binds to nucleic acid-containing self-antigens such as Sm/RNP and ribosomes, which are characteristic targets of autoantibodies in patients with systemic lupus erythematosus (SLE). Siglec-10 recognizes gangliosides, which are glycolipid antigens targeted by autoantibodies in patients with Guillain-Barré syndrome. Upon antigen binding through the B cell receptor (BCR), the Src-family kinase Lyn phosphorylates the cytoplasmic domains of these inhibitory receptors. These phosphorylation sites recruit and activate the tyrosine phosphatase SHP-1, which in turn inhibits BCR signaling. This suppression of BCR-mediated activation leads to self-tolerance in B cells specific for non-protein self-antigens and prevents the production of pathogenic autoantibodies.
B cell self-tolerance is generally regulated by T cell self-tolerance. Even if B cells recognize self-antigens, they are usually not activated in the absence of T cell help. However, when B cells recognize non-protein self-antigens or complex antigens containing non-protein components, the intrinsic activation signals from these non-protein antigens can potentially activate self-reactive B cells independently of T cell help, leading to autoantibody production. Therefore, self-tolerance of B cells against non-protein self-antigens—which cannot be regulated by T cell tolerance—may require direct suppression of B cell responses mediated by inhibitory receptors such as CD72 and Siglec-10.
Patients with SLE produce autoantibodies against various self-antigens, including nucleosomes, histones, DNA, Sm/RNP, and ribosomes. Similarly, CD72-deficient mice produce autoantibodies against a broad range of autoantigens. Our research group is currently elucidating the mechanism by which CD72 suppresses the production of autoantibodies against self-antigens beyond Sm/RNP and ribosomes. We are also developing novel therapeutic approaches for SLE by enhancing this regulatory mechanism.
Development of novel therapeutics for autoimmune diseases
Some B cells suppress immune responses by secreting inhibitory cytokines such as IL-10. These cells are referred to as regulatory B cells (Breg cells), and both the number and function of Bregs have been shown to be reduced in various autoimmune diseases. It has also been demonstrated that Bregs play a role in suppressing graft rejection. Although IL-6 and TLR ligands are known to enhance Breg cell function, they cannot be used as treatments for autoimmune diseases because they also promote inflammation.
We have identified a novel target molecule that enhances Breg cell function without inducing inflammation. We have demonstrated that antibodies against this molecule suppress the development of autoimmune diseases and graft rejection in mice. Based on these findings, we are currently developing new antibody-based therapeutics for autoimmune diseases.
In parallel, our research group is also developing novel therapeutic strategies for SLE that enhance the function of CD72.
Development of therapeutic vaccines as an alternative to therapeutic antibodies
Many of the groundbreaking therapeutic drugs developed in recent years are therapeutic antibodies. This is because antibodies can target molecules that are not accessible to small-molecule compounds. Conversely, it is extremely difficult to replace therapeutic antibodies with small molecules. However, due to the high cost associated with therapeutic antibodies, alternative approaches are needed.
One such approach involves immunizing patients with a portion of the target molecule as a vaccine, thereby inducing the patient’s immune system to produce antibodies against the target. These therapeutic vaccines may replicate the effects of therapeutic antibodies.
Our research group is developing fundamental technologies for therapeutic vaccines that can serve as alternatives to therapeutic antibodies. This technology has the potential to bring about a paradigm shift in disease treatment, comparable to the shift brought about by the advent of therapeutic antibodies.
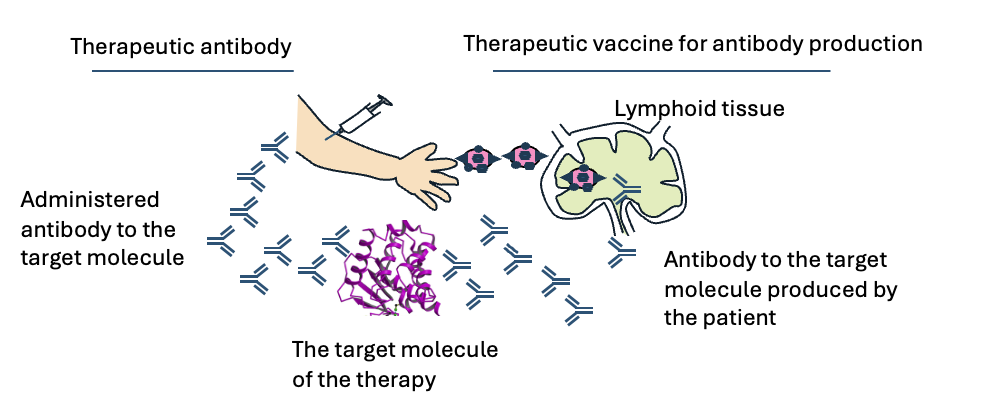
Figure 3. Antibody-producing therapeutic vaccines that can substitute for therapeutic antibodies
When administrated to patients, therapeutic antibodies bind to the target molecules and modulate their activities thereby generating therapeutic effects. Antibody-producing therapeutic vaccines contain a part of the therapeutic target molecules. When administrated to patients, these vaccines induce production of antibodies that bind to the target molecule and modulate their activity as well as therapeutic antibodies. As a result, antibody-producing therapeutic antibodies show similar therapeutic effects to therapeutic antibodies.